
Funny current. Neuronal firing patterns rely on the coordinated interplay of ion channels, mediating different aspects of how neurons fire and repolarize. In the early 70s, a strange phenomenon was observed: an ion current that became active when it shouldn’t. Subsequently named the h-current or funny current, it activates when neurons are at their resting potential, pulling them away from their resting membrane potential and helping with pace-making. HCN channels are the molecular equivalents of the h-current. In a recent publication, Houdayer and collaborators explore the phenotypic consequences of mutations in HCN2, one of the key components mediating the neuronal h-current. Here is a brief summary of the unusual clinical spectrum associated with variants in HCN2.
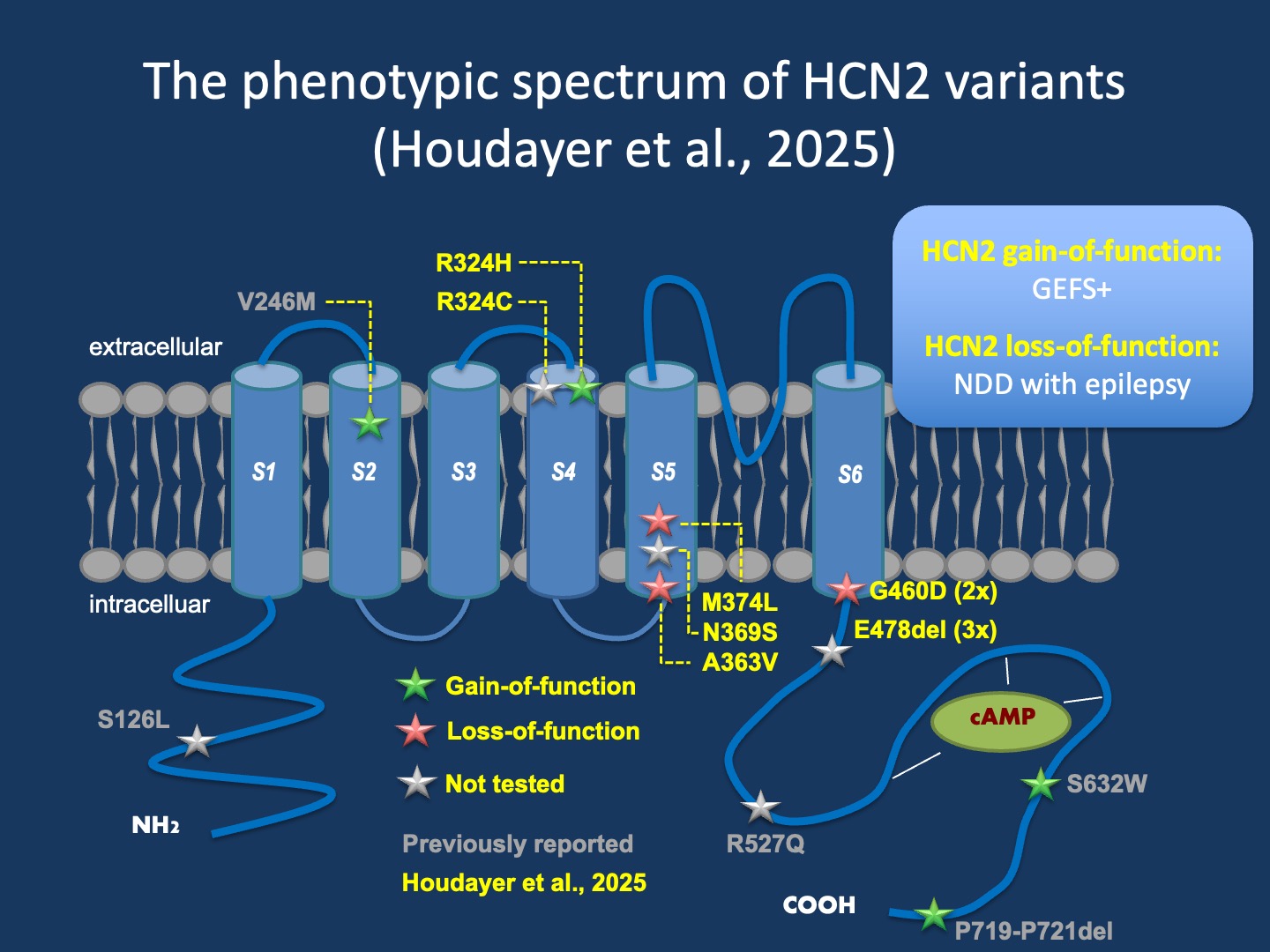
Figure 1. Disease-causing variants in HCN2 lead to distinct phenotypes depending on their functional consequences. HCN2 represents one of four genes encoding the hyperpolarization activated cyclic nucleotide (HCN) gated ion channels that play an important role in neuronal pace-making. Gain-of-function variants lead to epilepsy and febrile seizures with typical or borderline intellect, while loss-of-function variants lead to neurodevelopmental disorders, with epilepsy reported in less than 50% of individuals.
H-current. Over a decade ago, we reported on the discovery of de novo variants in HCN1 in developmental and epileptic encephalopathies (DEEs). The HCN1 gene is one of the components of the hyperpolarization activated cyclic nucleotide (HCN) gated ion channel, which functions as a pacemaker within neurons. While the initial report of de novo variants focused on DEE reminiscent of Dravet Syndrome, the spectrum of associated phenotypes was later expanded to include GEFS+. However, the predilection of fever-associated seizures stood out and remains mechanistically unexplained to date.
Enter HCN2. Both HCN1– and HCN2-related disorders are extremely rare, so it took me a while to realize that the genes are actually distinct. While HCN1 variants are an established cause of DEE, the evidence for HCN2 as a disease gene has been inconclusive. This changed with the recent publication by Houdayer and collaborators, who systematically assessed the clinical and phenotypic consequences of genetic variation in HCN2.
The phenotypic spectrum. Houdayer and collaborators reported on a cohort of both bi-allelic and mono-allelic variants. For the blog post, I focus only on mono-allelic variants, given that bi-allelic variants are generally thought to result in loss-of-function effects. The clinical spectrum of HCN2-related disorders is puzzling. It appears that both gain-of-function and loss-of-function variants result in distinct phenotypes that are counterintuitive when compared to other channelopathies.
Loss-of-function. Houdayer and collaborators report on 12 individuals with loss-of-function variants in their recent publication. Almost all individuals had intellectual disability in the moderate to severe range where information was provided. Epilepsy was reported in two-of individuals and typically started in infancy. This clinical presentation is somewhat different from many other complex neurodevelopmental disorders given the high seizure burden and age of onset. Clinical features were significantly more severe in the eight individuals with biallelic loss-of-function variants, suggesting a correlation between the severity of neurodevelopmental features and the degree of loss-of-function of HCN2.
Gain-of-function. In contrast, of the 18 individuals with gain-of-function variants, only a single individual had developmental differences. Additionally, while seizures were only found in 8/18 individuals for whom information was available, an additional eight individuals had febrile seizures. This phenotypic distribution is reminiscent of HCN1 and hints at an unusual connection between neuronal pace-making currents and sensitivity to fever-induced seizures.
What you need to know. In a recent publication, Houdayer and collaborators report on 21 individuals with HCN2 variants, including information from 15 unrelated families. The authors saw a clear genotype-phenotype correlation: gain-of-function variants caused fever-related seizures and few developmental differences, while loss-of-function variants typically resulted in neurodevelopmental disorders with moderate to severe intellectual disability concomitant with seizures in 67% of individuals. In summary, HCN2-related disorders are a relatively new entity. However, despite their recent discovery and limited number of reported individuals, HCN2-related disorders, present with vastly different phenotypes based on variant function, a crucial feature to understand in the near future for targeted therapies.