
Connections. Spectraplakins sit at the interface of architecture and motion inside a cell. They do not merely hold structures together but coordinate how the cytoskeleton rearranges itself when neurons migrate, polarize, and extend processes. One of the central spectraplakins is encoded by MACF1, a microtubule–actin crosslinking factor that couples microtubules to actin filaments and helps steer growing microtubules. This job requires multiple binding domains, flexibility, and scale. Spectraplakins are therefore large proteins encoded by enormous genes. A recent study examined how variants in MACF1 translate into human brain disease, and why seemingly similar variants may lead to surprisingly different neurodevelopmental outcomes. Here is why interpreting MACF1 variants is so complex.
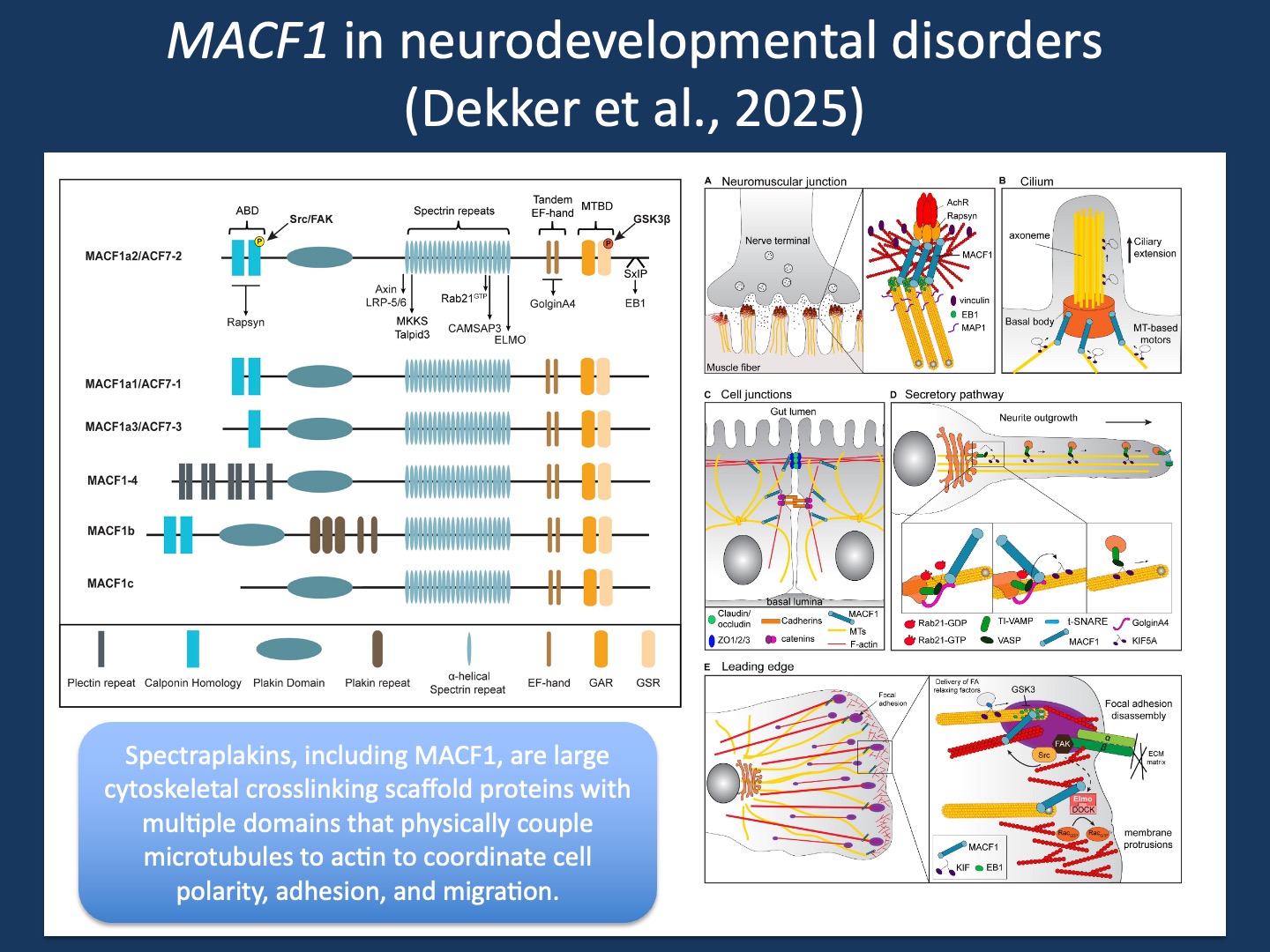
Figure 1. Spectraplakins, including MACF1, are large cytoskeletal scaffold proteins that crosslink actin filaments and microtubules to coordinate cellular architecture, polarity, and intracellular organization. This group of proteins has a modular architecture consisting of an N-terminal actin-binding domain, central spectrin-repeat rod regions, and C-terminal microtubule-binding motifs (left). These complex proteins have various functions in neurons, including regulation of neurite outgrowth, axon guidance, synapse organization, and intracellular trafficking (right). Figures from Cusseddu et al. (2021) under a Creative Commons CC BY license.
Connections. MACF1 biology is best understood as a bridge between cytoskeletal systems that are often discussed separately. Microtubules provide directionality, long-range transport, and structural exploration, while actin filaments provide force generation, membrane dynamics, and local control at the cortex and in growth cones. Spectraplakins connect these networks into one coordinated machine, anchoring microtubules within actin-rich compartments and stabilizing the geometry neurons depend on. This bridging function comes with a genetic price. MACF1 has 104 exons, and its transcript structure is complex enough that a single gene-level view can be misleading. In the recent AJHG study by Dekker and collaborators that we contributed to, five MACF1 isoforms were highlighted as expressed in the developing human brain, driven by alternative promoter usage and distinct first exons that converge on a shared downstream transcript architecture. In large genes, transcript reality becomes a practical challenge for curation and for clinical interpretation.
Concepts In ClinGen gene curation, we try to convert complexity into reproducible categories. The ClinGen framework evaluates gene–disease validity through structured evidence, forcing a clear separation between plausible stories and durable associations. Over the last few years, I have been the Co-Chair of the Epilepsy Gene Curation Expert Panel (GCEP) within the NIH-funded ClinGen consortium, a group that evaluates the evidence supporting gene–disease relationships in epilepsy and defines which genes truly qualify as established causes of disease. MACF1 illustrates a recurring tension in this work. It has a pLI close to 1, suggesting strong intolerance to loss-of-function variants and a relatively straightforward story of haploinsufficiency. But MACF1 is not straightforward. Different domains may cause disease through different mechanisms, and different transcripts may matter in different brain regions or developmental windows. This raises an uncomfortable but necessary question in curation: are we curating a single gene–disease relationship, or several overlapping relationships that share only a gene symbol.
MACF1 phenotypes. In the study by Dekker and collaborators, a cohort of 45 individuals shows that MACF1-related disease is better described as a spectrum than a single condition. One major pattern is a recognizable malformation phenotype including lissencephaly with brainstem and commissural defects, often summarized as LISDD, with multiple de novo missense variants clustering in the C-terminal microtubule-binding region (EF-hand and GAR modules). Functional assays support a distinct mechanism for these variants, consistent with increased microtubule binding rather than simple loss of function. Importantly, our study also describes biallelic loss of function early in the gene, including a homozygous exon 1 stop-gain, producing an LISDD spectrum phenotype that is linked to developmental isoform usage and promoter architecture.
Absent MACF1 phenotypes. What is not seen is equally informative. Despite MACF1 being a highly constrained gene, our study does not reveal a broad pattern of de novo protein-truncating variants across the gene, the signature one might expect for straightforward haploinsufficiency in the mold of STXBP1, SCN1A, or SYNGAP1. For biallelic variants beyond exon 1, the authors emphasize that there might be a broader neurodevelopmental phenotype, but the evidence is suggestive rather than definitive, with interpretive uncertainty amplified by the transcript complexity of MACF1. This shows that complex genes such as MACF1 can not only result in different phenotypes, but that these phenotypes may have different levels of evidence given the overall genomic noise in genes of this size.
What you need to know. MACF1 is a reminder that gene names can hide biological complexity. Spectraplakins such as MACF1 are cytoskeletal integrators that connect microtubules and actin and help cells translate polarity into movement and structure. The size of this task is reflected in the gene itself, with 104 exons and multiple transcripts with brain region-specific expression. Dekker and collaborators show that MACF1-associated disorders are unlikely to represent one unified gene–disease relationship, but rather several mechanistic pathways. Domain-specific variant clustering supports distinct mechanisms, including altered microtubule binding, while biallelic loss of function early in the gene expands the LISDD spectrum through a second route shaped by developmental transcript architecture. Even for a gene with strong constraint metrics, transcript context and mechanism can split the evidence into separate disease models, making careful phenotype definition as important as the variant itself.