
RNA. More than a decade ago, I remember reading an article arguing that we actually live in an RNA world. At the time, this felt a bit academic and not really relevant. Genetics was about sequencing and interpreting DNA. However, over the last few years, our ability to think in terms of RNA transcripts rather than DNA sequence has become increasingly relevant. When I teach trainees, I sometimes tell them: we do not care about genes. And then I pause, usually long enough to make people uncomfortable. Then I correct myself: we care about transcripts. In a recent publication, we assessed how a novel targeted long-read RNA sequencing approach can help with rare disease diagnosis. Here is what we found.
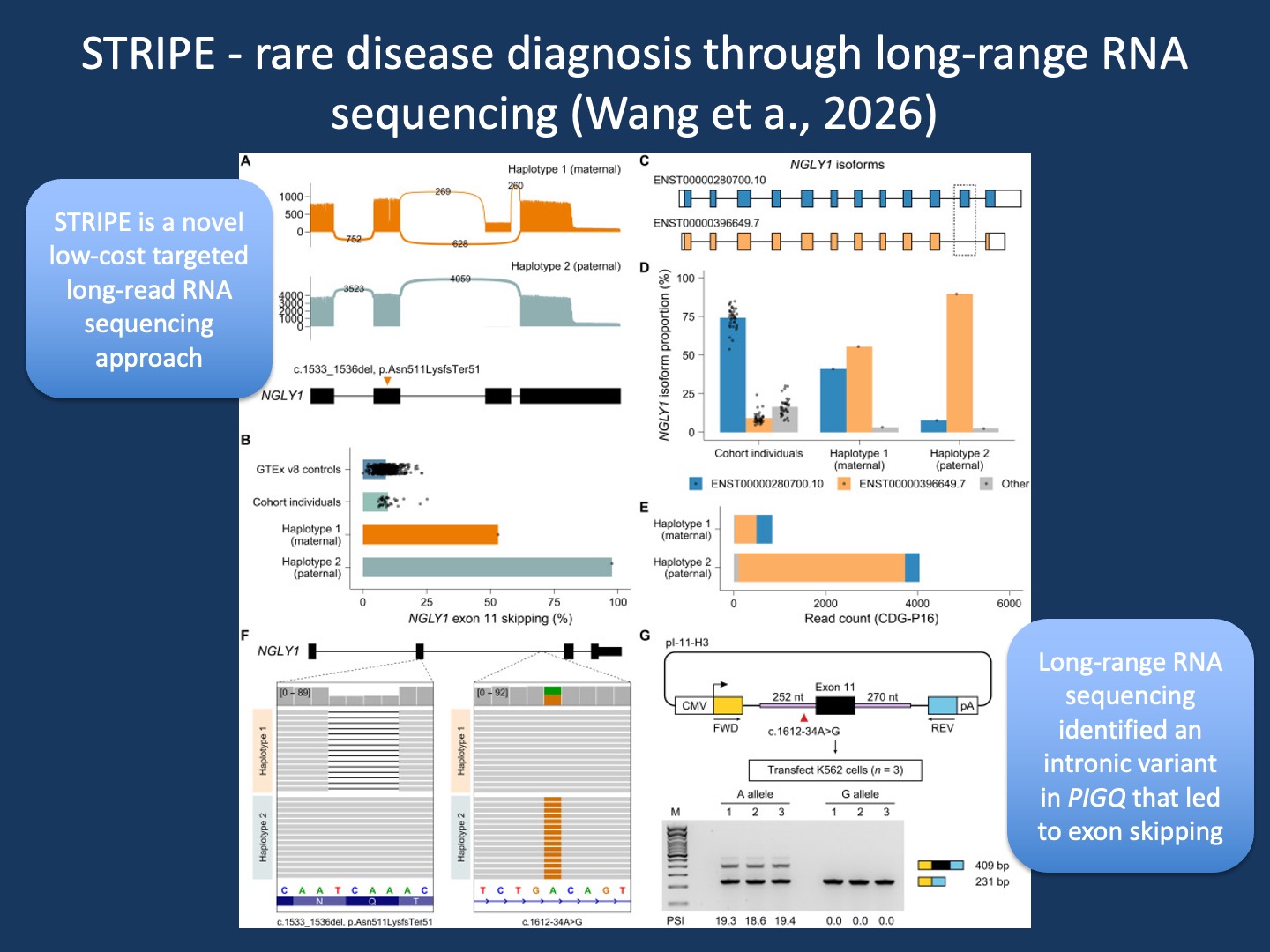
Figure. Transcript-level diagnosis in NGLY1-related disease using STRIPE. In one of the most instructive examples from our publication by Wang et al., STRIPE resolved the transcript consequences of two variants in an individual with NGLY1 deficiency. A maternally inherited frameshift variant and a paternally inherited deep intronic variant converged on a shared transcript-level mechanism: skipping of exon 11. The sashimi plots and isoform analyses show how the paternal allele shifts transcript architecture toward an exon-skipped isoform, a pattern that was largely absent in controls. Long-read sequencing made it possible to phase these changes by parental haplotype and to directly quantify the altered isoform proportions. Genomic long-read sequencing then identified the causative intronic variant, and minigene assays confirmed its effect on splicing. This example highlights why transcript diagnostics matter: the causative variant was not in the coding sequence, but its effect became immediately visible once we looked at RNA (adapted from Wang et al, 2026).
Epilepsy genetics has been spoiled. In epilepsy genetics, we still often think like gene people. Our field has, in some ways, been spoiled by simplicity. Many of the genes that built the epilepsy genetics field were quite generous to us: SCN1A, SCN2A, SCN8A, STXBP1. These genes had major isoforms where we could locate variants and think of a gene as a single instruction that generates a protein. For years, this allowed us to think in a relatively straightforward way. However, even in these major genes, there are examples where this simplicity does not hold true.
The poison exon lesson. Perhaps the clearest example is SCN1A. For years, SCN1A looked like a typical haploinsufficient disease gene. But then the poison exon story emerged. In brief, a significant portion of SCN1A transcription is shunted into a non-productive splicing isoform. This shunting keeps expression of full-length SCN1A low in early infancy and then switches over. This is when SCN1A haploinsufficiency and the clinical features of Dravet syndrome start to emerge. This biology is now used clinically to increase SCN1A transcription through antisense oligonucleotides (ASOs). SCN8A provides another example. The SCN8A gene uses an alternative exon 5 that includes de novo variants that had, at one point, been entirely missed in diagnostics.
Looking at transcripts. In our recent study in Science Advances by Wang and collaborators, we used a novel targeted long-read RNA sequencing approach called STRIPE. Long-read RNA sequencing is not new and allows us to look at entire transcripts rather than individual exons. However, STRIPE is different in an important way. Most RNA sequencing approaches, including long-read methods, either cast a broad net or require substantial amounts of input material. In contrast, STRIPE is targeted. It enriches specific transcripts of interest and sequences them deeply, allowing us to focus on disease-relevant genes with much greater resolution. This matters because rare splice events, alternative transcript architectures, and low-abundance isoforms can easily be missed in broader approaches. What makes STRIPE particularly useful is that the probe design is modular: once the exons of a gene set are defined, tiled capture probes can be generated in bulk at low cost and adapted to new panels without rebuilding the assay from scratch.
Examples. Some of the most instructive examples in our STRIPE paper come from genes that are already familiar to us in neurogenetics. For example, in a previously undiagnosed individual with early infantile epileptic encephalopathy, we identified PIGQ as the causative gene. STRIPE showed how a splice-altering variant in PIGQ generated abnormal transcript architectures that would have been difficult to reconstruct from short reads alone. In another individual, we identified a paternally inherited intronic variant in NGLY1 that led to skipping of an entire exon, a variant that would likely not have been recognized as causative without directly observing the altered splicing patterns.
Eighteen years later. I started my career in epilepsy genetics with a small gene expression study in absence epilepsy, trying to understand whether transcriptional signatures could point us toward disease biology. At the time, the idea felt right, but the tools were not there yet. We could measure expression, but not transcripts in the way that matters now. It was expensive, low-scale, and difficult to analyze, generating signals that were often hard to connect back to actual transcript architecture. Looking back, STRIPE feels like the technology I wish we had then. Its advantages are surprisingly practical. First, it is low-cost, which matters because transcript-level diagnostics only become useful if they can scale beyond individual research projects. Second, it is long-read, meaning we can observe full transcripts directly instead of reconstructing them from fragments. And third, it is deep. Because STRIPE is targeted, sequencing depth allows us to detect rare or unexpected transcript isoforms that broader transcriptome approaches would simply miss.
What you need to know. The main lesson from STRIPE is somewhat simple: if we want to understand what a variant does, we often need to look at the transcript. For a long time, epilepsy genetics could operate with a relatively gene-centered view because many of our major disease genes had transcript structures that seemed straightforward. But the examples of SCN1A poison exons, alternative exons in SCN8A, and splice-disrupting variants in genes such as PIGQ and NGLY1 remind us that the biology is often much more complex. STRIPE makes transcript-level analysis practical by combining low cost, deep sequencing, and long-read resolution. If the first phase of epilepsy genetics was about finding genes, the next phase may be about understanding the transcripts they actually produce.