
Signal. I admit that the title of this blog post is somewhat misleading, but please bear with me. Yes, GLP-1 receptor agonists have very little to do with epilepsy, but there is a larger story behind this. In a recent study, nearly 28,000 people taking GLP-1 receptor agonists answered a seemingly simple question: how much weight did you lose, and how bad were the side effects? This simple survey, coupled with genetic data, produced one of the cleanest pharmacogenomic signals seen in recent years. But it also emphasized that the genetics of treatment are often not the genetics of disease, and that matters far beyond obesity and weight loss. Here is why this should make us rethink pharmacogenomics in epilepsy.
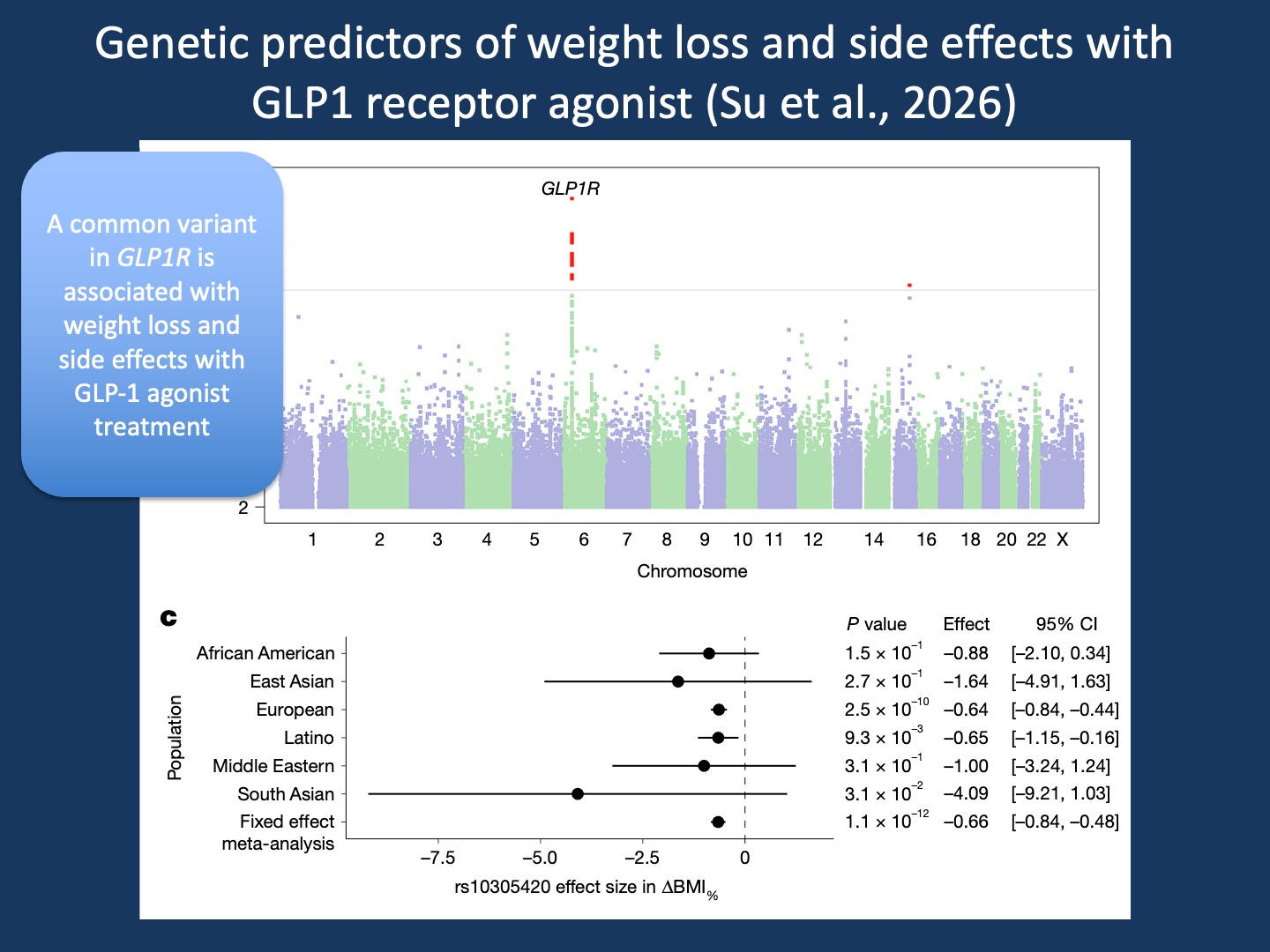
Figure. In the study by Su et al. in Nature of nearly 28,000 individuals treated with GLP-1 receptor agonists, a genome-wide association study identified a common variant in GLP1R (rs10305420) associated with greater weight loss. The Manhattan plot (top) shows a clear genome-wide signal centered on GLP1R, the direct molecular target of semaglutide and tirzepatide. The forest plot (bottom) demonstrates that the effect is directionally consistent across ancestral groups, with the strongest signal in European participants and a significant fixed-effect meta-analysis across cohorts. Importantly, this variant is not primarily associated with obesity risk itself. Instead, it predicts response to treatment, illustrating a central concept in pharmacogenomics: the genetics of drug response may be distinct from the genetics of disease. This distinction is the starting point for understanding why pharmacogenomics in epilepsy remains so difficult.
GLP-1 agonists. Glucagon-like peptide 1 (GLP-1) receptor agonists have changed the landscape of obesity treatment in a remarkably short time. Drugs such as semaglutide and tirzepatide mimic incretin hormones, signals released after food intake that stimulate insulin secretion. These peptide hormones slow gastric emptying and reduce appetite through effects in the central nervous system. What initially began as a treatment for diabetes has rapidly expanded into obesity care, with an estimated one in eight adults in the United States having used a GLP-1 agonist. That is a striking number for a drug class that has only become widely used in the last few years and was barely part of the public consciousness before. But the response has been variable and understanding how genetic factors shape drug response is exactly the task of pharmacogenomics.
Su et al. In a recent study in Nature, Su and colleagues examined nearly 28,000 individuals with genomic data taking semaglutide or tirzepatide. The authors surveyed weight loss and side effects and performed a genome-wide association study (GWAS). They identified a missense variant in GLP1R, the drug target itself, associated with greater weight loss, and additional variants in GLP1R and GIPR associated with nausea and vomiting. At first glance, this sounds almost trivial: response to medications such as semaglutide or tirzepatide is linked to genetic variation in the biological pathway itself. But the implications are much more surprising than they seem at first.
The wrong comparison. If you know very little about GLP-1 agonists, the obvious instinct is to think about obesity genetics. We have well-established polygenic risk scores for BMI, obesity susceptibility, and metabolic syndrome, and it would be easy to assume that the genetics of weight loss should overlap with the genetics of weight gain. But that is not what this study found. The GLP1R variant associated with semaglutide and tirzepatide response is not primarily an obesity-risk variant. It does not explain who becomes obese. Instead, it helps explain who responds to treatment. That distinction is easy to miss, but it may be the most important lesson from the study by Su and collaborators. Disease genetics tells us why a condition happens, whereas pharmacogenomics tells us what happens when the underlying biology is perturbed by treatment. And this is where the link to epilepsy genetics comes into play.
The hidden genome. It may be useful to think of pharmacogenomics as uncovering a hidden genome: variation that exists in all of us, but remains functionally quiet until a drug turns the system on. Common variants in HLA-B, for example, can make people many-fold more susceptible to severe drug reactions with abacavir, allopurinol, or carbamazepine, which we wrote about previously. In the study by Su and colleagues, the signal sits in the signal peptide of GLP1R, likely affecting receptor trafficking rather than obesity itself. Under ordinary physiology, the effect of this variant may be subtle. But when exposed to non-physiological doses of semaglutide, it becomes visible. This framing helps explain why pharmacogenomics often feels disconnected from disease genetics. We are not looking at baseline biology. We are looking at biology under intervention, when previously silent genetic variation becomes unmasked.
Epilepsy has a different problem. Coming back to epilepsy pharmacogenomics, a recent systematic review by Barguilla and collaborators reveals a striking contrast. Across 168 studies, the field has accumulated many candidate associations, but very few robust signals that have held up over time. And the strongest findings are not about positive drug response, but toxicity. As mentioned previously, HLA alleles such as HLA-B*15:02 predict severe cutaneous reactions to carbamazepine and phenytoin, and CYP2C9 variants predict phenytoin toxicity. But when it comes to the question of whether somebody responds to a given anti-seizure medication, the answers remain weak and only few associations have been replicated. In epilepsy, we are much better at predicting side effects than drug response.
When biology is simple. In a way, this contrast makes biological sense. Toxicity is mechanistically cleaner. An HLA molecule presents a peptide and triggers an immune reaction. Drug metabolism changes plasma levels. These are relatively direct mechanisms. Seizure response is different. It involves network excitability, compensation, developmental timing, and systems-level effects rather than a clean molecular mechanism. This may help explain why epilepsy pharmacogenomics has struggled.
The sodium channel exception. There are exceptions, and in a way they prove the rule. In epilepsy, sodium channel disorders remain the clearest example of mechanistic pharmacogenomics. Gain-of-function variants in SCN2A or SCN8A may respond well to sodium channel blockers. Loss-of-function SCN1A disorders such as Dravet syndrome may worsen with them. This makes biological sense because the causative gene and the treatment mechanism overlap. But this is unusual. For most anti-seizure medications, the molecular target is not the causative genetic etiology.
The wrong assumption in precision medicine. There is an implicit belief in rare disease medicine that once we know the causative gene, treatment decisions should follow naturally. But more often than not, drug response in genetic epilepsies remains complicated. Individuals with the same causative variant in genes such as STXBP1, SYNGAP1, DNM1, or CDKL5 may respond very differently to the same anti-seizure medication. Shared disease biology does not necessarily imply shared pharmacologic response. The GLP-1 paper by Su and colleagues reminds us that there may be a second genome of treatment, separate from the genome of disease.
What you need to know. The recent study in Nature on GLP-1 agonists and a recent systematic review on epilepsy pharmacogenomics point to the same conclusion from opposite directions. In obesity, pharmacogenomics is beginning to uncover strong and biologically plausible treatment-response variants that are largely separate from disease susceptibility. In epilepsy, outside of rare mechanistic exceptions such as sodium channel disorders, we still know remarkably little about why anti-seizure medications work in some people and fail in others. Disease genetics and treatment genetics are not the same thing. For precision medicine in epilepsy, we need both.