
Recognition. There are moments when a new disease gene is reported and immediately feels important. And then there are moments when the same gene appears in three independent papers in the same issue of Nature Genetics. That is what happened with RNU2-2. In three parallel studies, independent groups essentially came to the same conclusion: biallelic variants in RNU2-2 are an unusually frequent cause of neurodevelopmental disorders and typically present as developmental and epileptic encephalopathies (DEEs). Taken together, these papers expand the spliceosome story that began with RNU4-2 in 2024. Here is what I learned about the recently identified recessive RNU2-2 disorders.
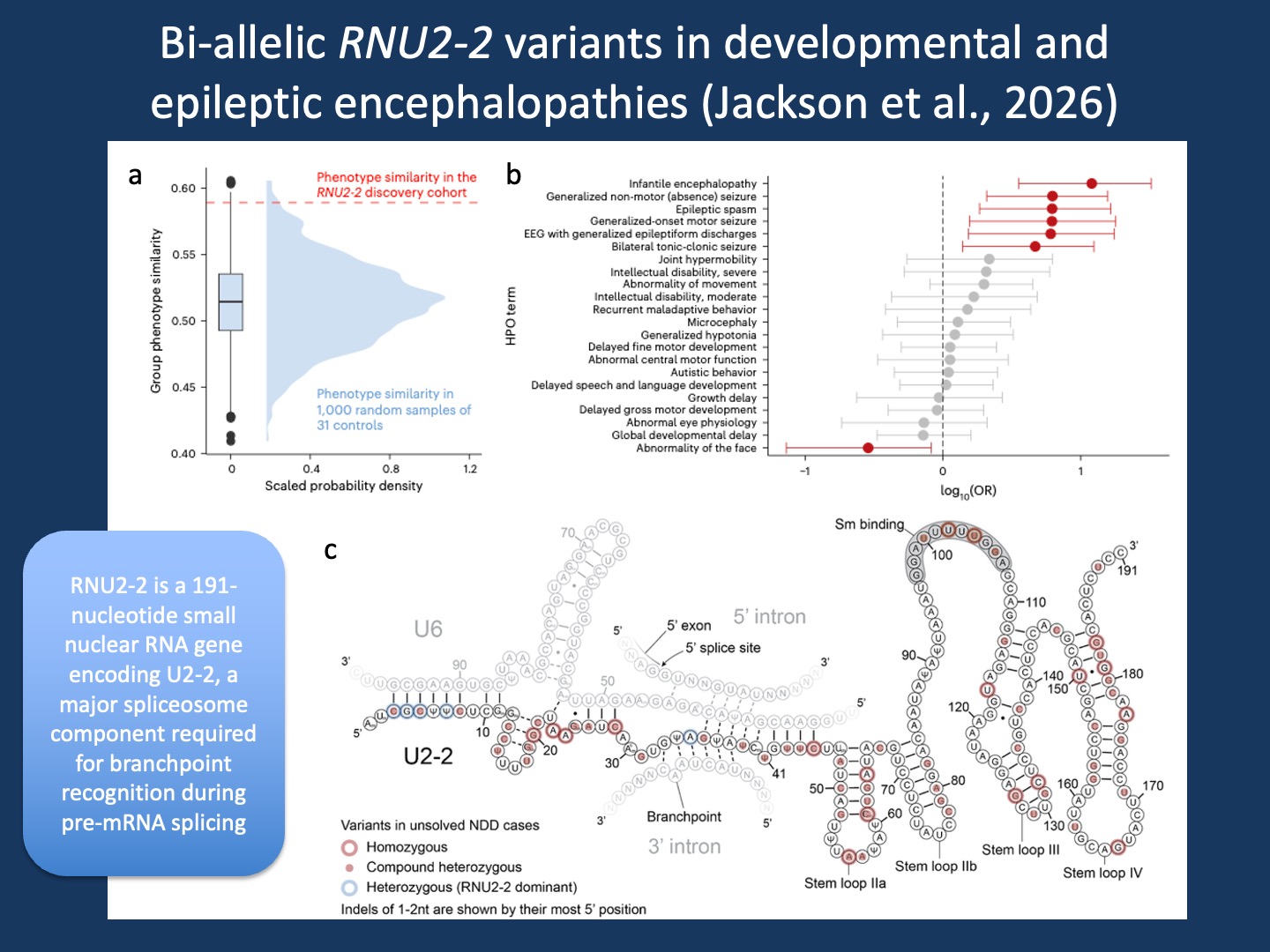
Figure. Bi-allelic RNU2-2 variants define a developmental and epileptic encephalopathy. Adapted from Jackson et al., 2026 under the Creative Commons Attribution 4.0 International License. Panel A shows that individuals with candidate bi-allelic RNU2-2 variants are more phenotypically similar to each other than expected from random groups of individuals with neurodevelopmental disorders in the 100,000 Genomes Project. Panel B shows the HPO enrichment analysis that defines the clinical signal. Panel C shows the proposed secondary structure of U2-2 in complex with U6, the 5′ splice site, and the splicing branchpoint, based on the four-helix U2/U6 structure. The original article is open access and licensed under CC BY 4.0, which permits use, sharing, adaptation, distribution, and reproduction with appropriate credit to the authors and source, a link to the license, and indication of changes.
The spliceosome connection. When RNU4-2 first entered the field, it changed how we thought about the spliceosome. Until then, the spliceosome often felt like an important piece of the cellular biology, but not necessarily the place where one would expect recurrent disease mechanisms to emerge. The discovery of RNU4-2-related neurodevelopmental disease challenged that assumption. RNU genes encode small nuclear RNAs, the RNA components of the spliceosome that help remove introns from precursor messenger RNA before it becomes mature mRNA. In the case of RNU2-2 and RNU4-2, these RNAs are directly involved in splicing, meaning that even subtle disruption can alter gene expression across hundreds of transcripts. What makes RNU2-2 particularly interesting is that it now appears to include different inheritance models. We already knew that heterozygous RNU2-2 de novo variants can cause dominant neurodevelopmental disease. These three new papers now establish that biallelic variants cause a recessive DEE, and in some cohorts the recessive form may even be more frequent than the dominant one. This is really highly unusual for an epilepsy gene.
Three papers, three perspectives. One reason the RNU2-2 feels unusually complete is that each publication contributes a different layer of evidence. The paper by Leitão and colleagues takes the broadest view, systematically screening 200 snRNA genes across more than 34,000 individuals with rare disorders and identifying RNU2-2 as one of the strongest recurrent signals. This framework is important because it suggests an interesting gradient of variant impact. Some variants may be sufficiently disruptive to cause dominant disease, while other variants function enough that a second variant is required. In contrast, the paper by Greene and colleagues approaches the question through statistical genetics, using Bayesian association to establish recessive inheritance with strong confidence. But the paper by Jackon and colleagues was the one that actually caught my eye as it used an HPO-based similarity analysis to assess what the recessive RNU2-2 phenotypes looks like.
Phenotypic similarity. Human Phenotype Ontology (HPO)-based phenotypic similarity analysis is an approach we have worked with extensively over the last years. At first glance, this can sound abstract when we try to convert clinical complexity into ontology terms and computational distances. But over time it has become increasingly clear that this approach can reveal patterns at scale that are otherwise difficult to recognize. In our own work, beginning with AP2M1 in 2019, we used phenotypic similarity across unrelated individuals as part of the evidence supporting a novel DEE gene. Since then, this idea has developed into something broader. In our recent work led by Shiva Ganesan that is currently accessible as a preprint, we found that phenotypic similarity can systematically identify gene-specific signatures across highly heterogeneous neurodevelopmental disorders across more than 11,000 individuals with trio exome sequencing. This was the transition we wrote about in 2020 when discussing the “phenotype era” in epilepsy genetics: we were finally able to think about phenotypes systematically in the same way we previously did with genotypes.
RNU2-2 similarity. The Jackon paper illustrates the idea of computational phenotyping particularly well. Their HPO enrichment analysis showed that individuals with biallelic RNU2-2 variants did not simply share broad neurodevelopmental features. Instead, they clustered around a developmental and epileptic encephalopathy (DEE) phenotype, including infantile encephalopathy, EEG abnormalities, and generalized seizure phenotypes as enriched features. That may feel obvious in hindsight, once the gene is already established. And it does not yet have the full delineation of the phenotype that we would like to see in a case series. But identifying a coherent DEE signal within a large heterogeneous neurodevelopmental cohort is really challenging. That is where HPO-based phenotypic similarity becomes powerful. But once the phenotype is examined systematically, the shared structure becomes difficult to ignore. Of note, the Greene paper arrives at a similar conclusion from a different angle, identifying seizure-related HPO terms as statistically enriched among affected individuals. Both publications highlight that it is possible to obtain an initial insight into the full spectrum of phenotypes using existing datasets. And this ability significantly accelerated the discovery of recessive RNU2-2 disorders and the strong link to DEE phenotypes.
What you need to know. The three new Nature Genetics papers on RNU2-2 establish one of the most frequent recessive DEEs identified so far. For me, the strongest thread connecting these papers is the phenotypic analysis using HPO terms. At the same time, RNU2-2 extends the spliceosome story that began with RNU4-2, reinforcing the idea that core RNA biology is increasingly central to neurodevelopmental disease. And when re-analyzing unsolved DEE cohorts, RNU2-2 now feels less like an unusual candidate and more like a gene that should already be part of the conversation.